Thermally activated dynamic gating in metal–organic frameworks
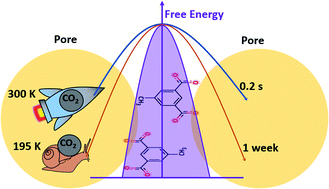
In unraveling the mechanism of kinetically governed gas adsorption in flexible metal–organic frameworks (MOFs), the careful employment of advanced computational tools can play a vital complementary role to experiments. Herein, we report the computational study of CO2 adsorption in a flexible MOF, CID-Me ([Zn(5-methylisophthalate)(bipyridine)]n), which showed an unusual higher gas uptake at higher temperatures. Density functional theory calculations yield a CO2 binding energy of around 11 kcal mol−1, with the availability of two equivalent CO2 binding sites per pore. Well-tempered metadynamics based molecular dynamics simulations are employed to delineate the diffusion mechanism of CO2 between pores which is facilitated by a thermally activated dynamic gating process. The minimum free energy path for CO2 diffusion exhibits a barrier of more than 16 kcal mol−1, which underpins the increased gas adsorption at higher temperatures. Molecular level insights on the libration of linkers gained here are used to elaborate on the appropriate design of materials which can adsorb more gas upon heating.